
Molecular Dynamics (MD) simulation is a computational method based on statistical mechanics and thermodynamics theory to simulate the interactions and behaviours of various atoms and molecules. MD simulations coupled with the Reactive Force Field (ReaxFF) allow the consideration of disassociation and formation of chemical bonds, which can be used to study pyrolysis breakdown of a material. ReaxFF provides more in-depth understanding into the mechanisms of the pyrolysis process because chemical reactions can be observed at the molecular level. Atomistic-scale computational techniques provide a powerful means for exploring, developing and optimising promising properties of novel materials.
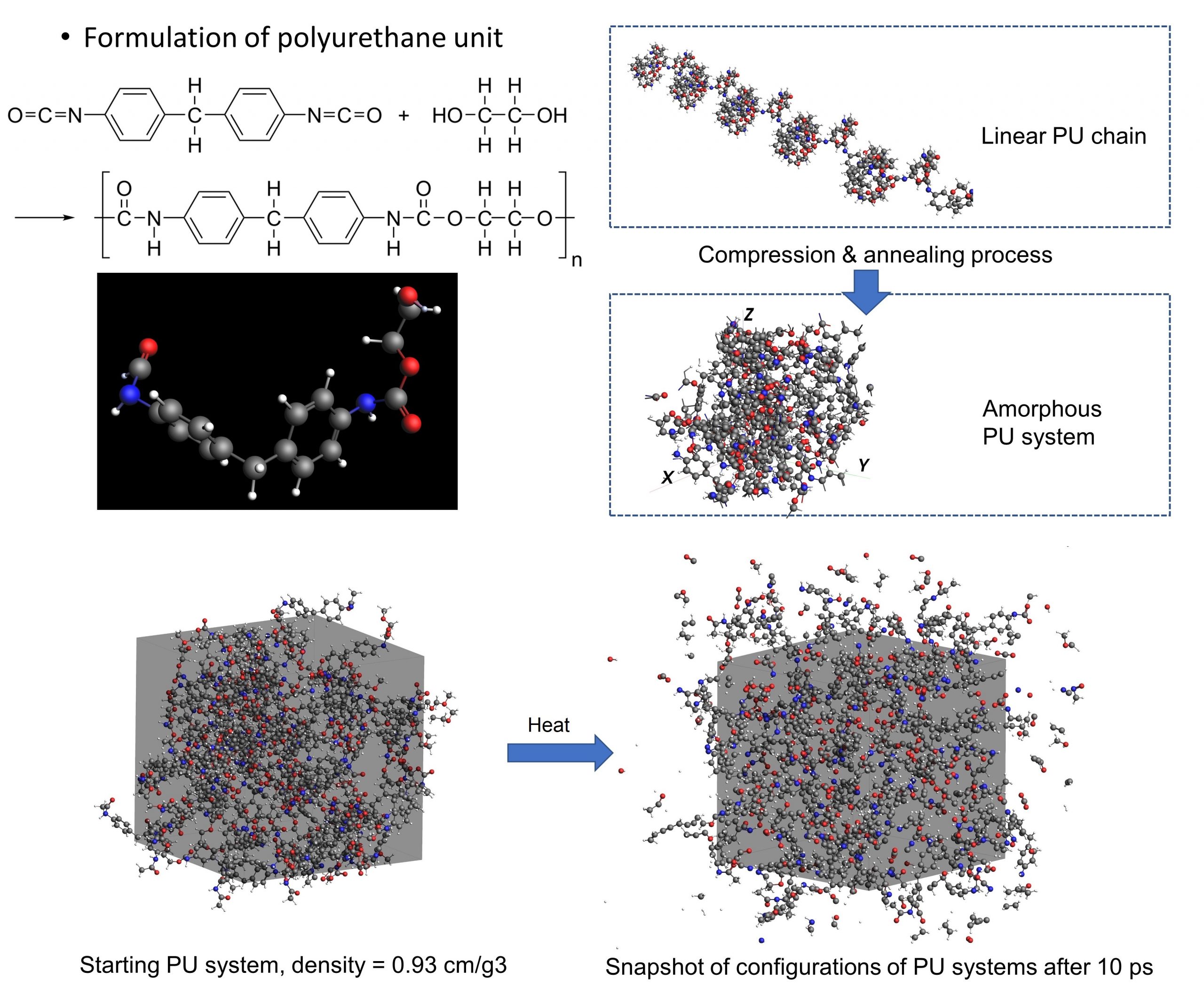
In summary, MD removes the need for complex multiphase pyrolysis and charring models which require substantial prior knowledge on material characterisation. It also serves as effective computational experiments to analyse material properties and predict mechanical responses. Furthermore, this technique has great potential for CFD pyrolysis modelling applications by providing the essential precursors of combustible fuel gases in combustion models to significantly enhance the reliability of toxic gas, charring, and smoke particulate predictions.

